AI-Based Drug Discovery
Our group develops and validates new computational methods to predict and model molecular recognition processes related to drug design with particular focus on the integration of deep learning and physicochemical modeling. In addition to high-content screening and de novo design, the main focus is a completely new physicochemistry-guided AI platform for structure-based drug design. Examples of recent developments in our group for AI-based drug discovery methods are:
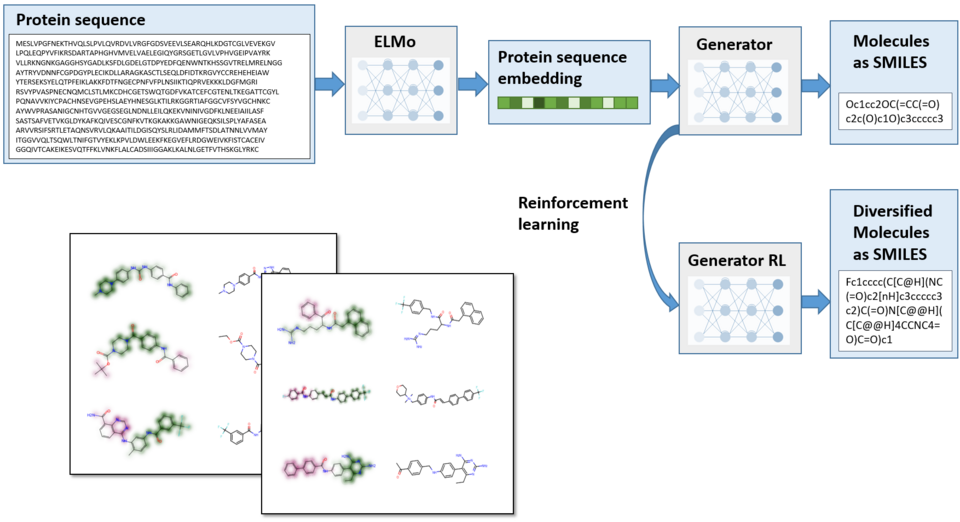
Protein-specific de novo design
To design focused compound libraries with high potential for binding to a particular target protein, we recently developed a de novo molecular design protocol combining transformer and recurrent neural network models. Using only sequence information about the target protein, this model allows to enrich libraries with compounds that have increased probabilities for binding to this specific macromolecule.
Ghanbarpour, A.; Lill, M.A. Seq2Mol: Automatic design of de novo molecules conditioned by the target protein sequences through deep neural networks arXiv:2010.15900, 2020.
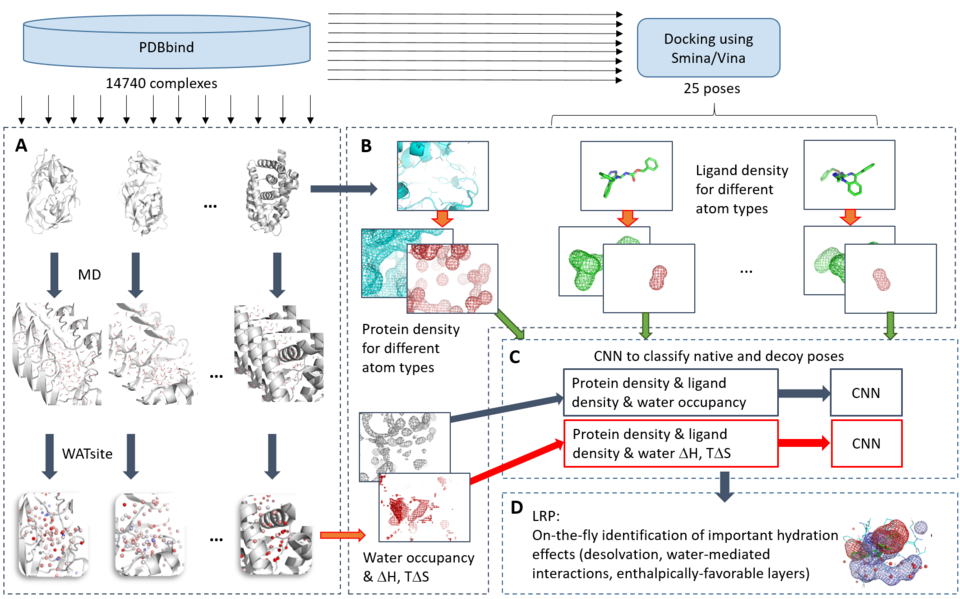
Accurate binding-pose prediction using physics-based AI
Accurate and efficient prediction of protein-ligand interactions has been a long-lasting dream of practitioners in drug discovery. The insufficient treatment of hydration is widely recognized to be a major limitation for accurate protein-ligand scoring. Using an integration of molecular dynamics simulations on thousands of protein structures with novel big-data analytics based on convolutional neural networks and deep Taylor decomposition, we consistently identify here three different patterns of hydration to be essential for protein-ligand interactions. In addition to desolvation and water-mediated interactions, the formation of enthalpically favorable networks of first-shell water molecules around solvent-exposed ligand moieties is identified to be essential for protein-ligand binding. Despite being currently neglected in drug discovery, this hydration phenomenon could lead to new avenues in optimizing the free energy of ligand binding. Application of deep neural networks incorporating hydration to docking allowed us to reduce the error rate of standard docking (around 40 %) to about 10 %, making it the most accurate pose ranking method published to date.
Mahmoud, A.H.; Masters, M.R.; Yang, Y.; Lill, M.A. Elucidating the multiple roles of hydration for accurate protein-ligand binding prediction via deep learning. Commun. Chem. 3, 2020, 19.
WATsite-on-the-fly
We also developed deep neural network models to compute hydration density and thermodynamic profiles, thus replacing time consuming MD simulations for hydration site prediction. Using a combination of MIFs, spherical-harmonics-based featurization and neural network models, we were able to generate the converged thermodynamic state of dynamic water molecules in the heterogeneous protein environment based solely on the information of the static protein structure. The applicability of our machine learning methods to predict the hydration information is demonstrated in two different studies, the qualitative analysis and quantitative prediction of structure-activity relationships, and the prediction of protein-ligand binding modes.
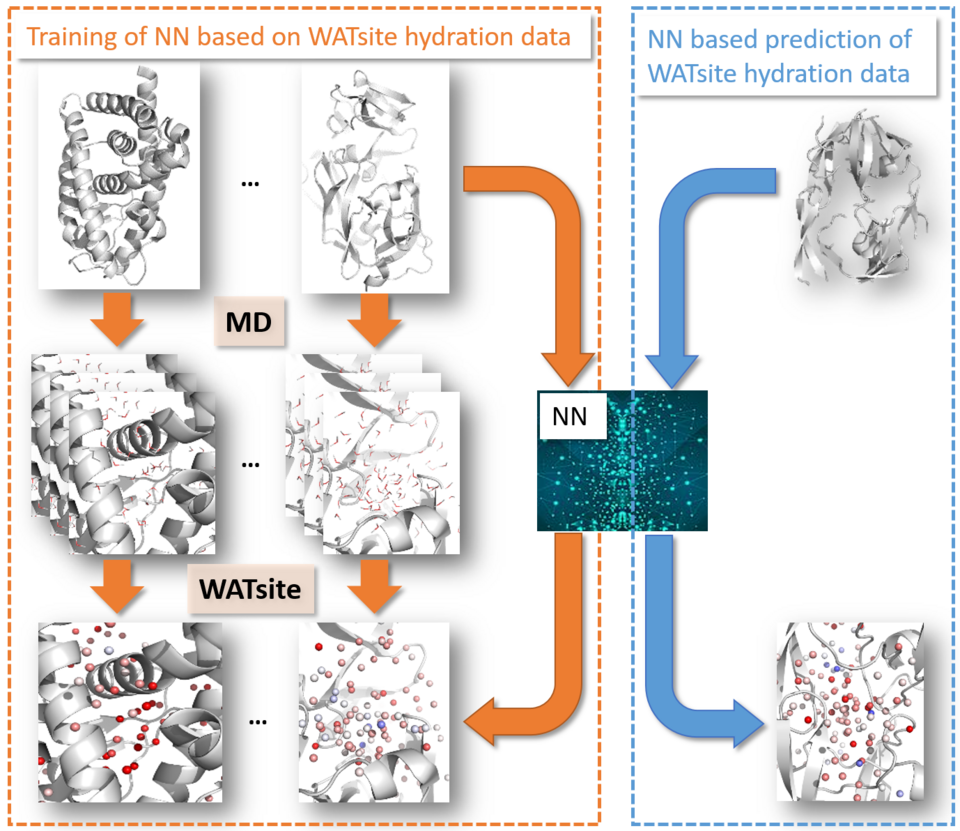
Ghanbarpour, A.; Mahmoud, A.H.; Lill, M.A. Instantaneous generation of protein hydration properties from static structures. Commun. Chem. 3, 2020, 188.
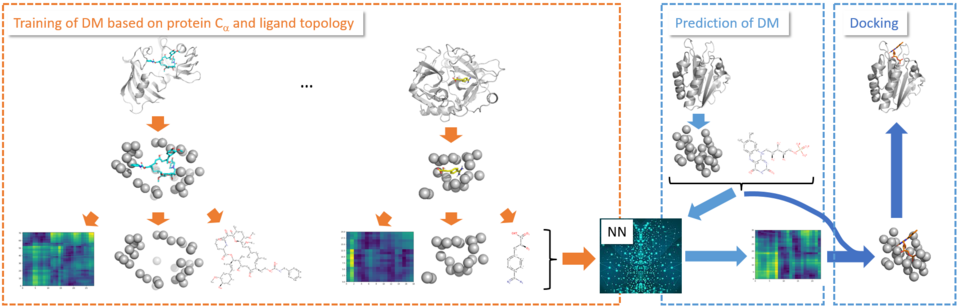
Coarse-grained docking using graph attention neural networks
We developed a completely different approach to the problem of flexible molecular docking. In the concept PoseNetDiMa pose generation with a flexible protein is performed based on graph convolutional neural networks, with significantly improved pose generation compared to standard flexible docking methods. PoseNetDiMa relies on a coarse-grained representation of the protein, and is trained to predict the distance matrix between ligand heavy atoms and centroids of binding site residues. This distance matrix is translated into poses of the ligand in the binding site of the protein, thus overcoming any time-consuming sampling of protein-ligand configurations.
Mahmoud, A.H.; Lill, J.F.; Lill, M.A. Graph-convolution neural network-based flexible docking utilizing coarse-grained distance matrix. arXiv:2008.12027, 2020.