Protein-Ligand Interactions
We have developed an arsenal of different computational methods to model molecular recognition events, predicting (1) if, (2) in which conformation and (3) how strong a molecule can bind to a protein. My group has addressed all three questions trying to most realistically and efficiently model all aspects of the protein-ligand system. My group focuses thereby on two important aspects of molecular recognition:
Solvation
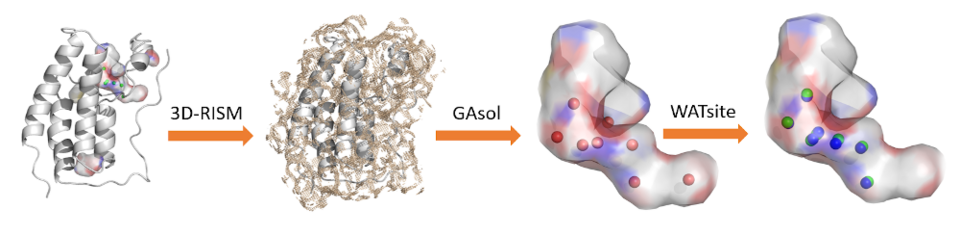
Water molecules in the binding site of proteins are important for molecular recognition as they can either bridge interactions between protein and ligand, or can be displaced by the bound ligand. Both mechanisms contribute enthalpically and entropically to the binding free energy, often driving the binding process. To elucidate the thermodynamic profile of individual water molecules and their potential contribution to ligand binding, we have developed a hydration site analysis program WATsite together with an easy-to-use graphical user interface based on PyMOL. WATsite identifies hydration sites from a molecular dynamics simulation trajectory with explicit water molecules. The free energy profile of each hydration site is estimated by computing the enthalpy and entropy of the water molecule occupying a hydration site throughout the simulation.
Hu, B.; Lill, M.A. WATsite: hydration site prediction program with PyMOL interface. J. Comput. Chem. 35, 2014, 1255-1260.
Yang, Y.; Hu, B.; Lill, M.A. WATsite2.0 with PyMOL Plugin: Hydration Site Prediction and Visualization. Methods Mol Biol. 1611, 2017, 123-134.
Yang, Y., Abdallah, A.H.A., Lill, M.A. Calculation of Thermodynamic Properties of Bound Water Molecules. Methods Mol. Biol. 1762, 2018, 389-402.
Masters, M. R., Mahmoud, A. H., Yang, Y., Lill, M. A. Efficient and Accurate Hydration Site Profiling for Enclosed Binding Sites. J. Chem. Inf. Model. 58, 2018, 2183-2188.
All previous hydration-site related studies have neglected the conformational change of the protein upon ligand binding. The use of hydration site information from a single protein conformation, however, is most likely incorrect for ligand binding. Therefore, we developed two methods to dissect the changes in location and free energy of hydration sites upon protein conformational change.
Yang, Y.; Hu, B.; Lill, M.A. Analysis of factors influencing hydration site prediction based on molecular dynamics simulations. J. Chem. Inf. Model. 54, 2014, 2987-2995.
Yang, Y.; Lill, M.A. Dissecting the Influence of Protein Flexibility on the Location and Thermodynamic Profile of Explicit Water Molecules in Protein-Ligand Binding. J. Chem. Theory Comput. 12, 2016, 4578-92.
Using WATsite on thousands of proteins in conjunction with modern deep learning approaches allowed the successful modeling of hydration during scoring of protein-ligand binding poses. This on-the-fly inclusion of hydration information resulted in unprecedented accuracy in binding pose prediction. Big-data analytics based on relevance deduced from the trained neural network revealed that the correct prediction of binding poses depends on three essential pillars of hydration, i.e. water-mediated interactions, desolvation, and enthalpically stable water layers around the bound ligand. The latter form of hydration may open new avenues for optimizing ligands for diverse protein targets.
Mahmoud, A. H.; Masters, M. R.; Yang, Y.; Lill, M. A. Elucidating the Multiple Roles of Hydration in Protein-Ligand Binding via Layerwise Relevance Propagation and Big Data Analytics. ChemRxiv, 2019.
Protein Flexibility & Dynamics
Flexibility and dynamics are protein characteristics that are essential in the process of molecular recognition. Studies from our and other groups have demonstrated that protein conformational changes are frequently induced by the bound ligand. To sample protein conformations that are relevant for binding structurally diverse ligands, we first introduced the methodology Limoc, the new concept of a hypothetical ‘ligand model’: a virtual ligand that binds to the protein and dynamically changes its shape and properties during sampling of protein conformations. In this method, MD simulations are performed with a dynamically changing set of restrained functional groups in the binding site of the protein, essentially representing a large hypothetical ensemble of different chemical species binding to the same protein. Beginning with an individual apo or holo structure of the protein, the ligand-model approach can be used to derive an ensemble of protein structures (EPS) used for identifying energetically feasible protein-ligand configurations. This EPS is used to represent alternative, pre-generated protein conformations in docking for efficient sampling of feasible protein-ligand configurations.
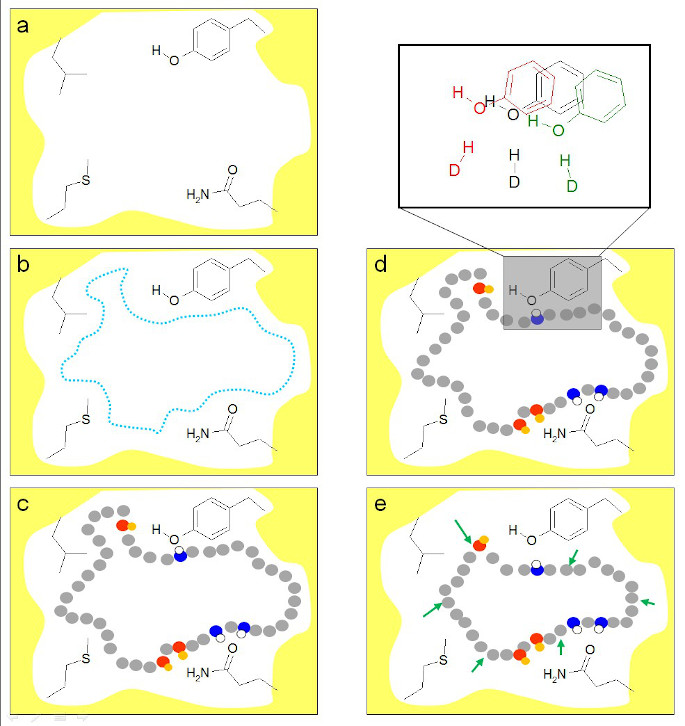
Xu, M.; Lill, M.A. Utilizing experimental data for reducing ensemble size in flexible-protein docking. J. Chem. Inf. Model., 52, 2012, 187-198.
Xu, M.; Lill, M.A. Significant enhancement of docking sensitivity using implicit ligand sampling. J. Chem. Inf. Model., 51, 2011, 693-706.
Recently, we developed new cosolvent simulation concepts to simulate protein conformational changes induced by the binding of different ligands. Cosolvent molecular dynamics (MD) simulations perform MD simulations of the protein in explicit water mixed with cosolvent molecules that represent functional groups of ligands potentially binding to the protein. The competition between different probes and water molecules allows the identification of the energetic preference of functional groups in different binding site moieties including enthalpic and entropic contributions. Cosolvent MD simulations have recently been applied to a variety of different questions in structure-based drug design but still have significant shortcomings. Among those issues is the limited chemical diversity of probe molecules ignoring the chemical context of the pharmacophoric feature represented by a probe. In our novel cosolvent MD simulation method, based on the λ-dynamics simulation concept, a significant increase in chemical diversity of functional groups investigated during cosolvent simulations is made possible.
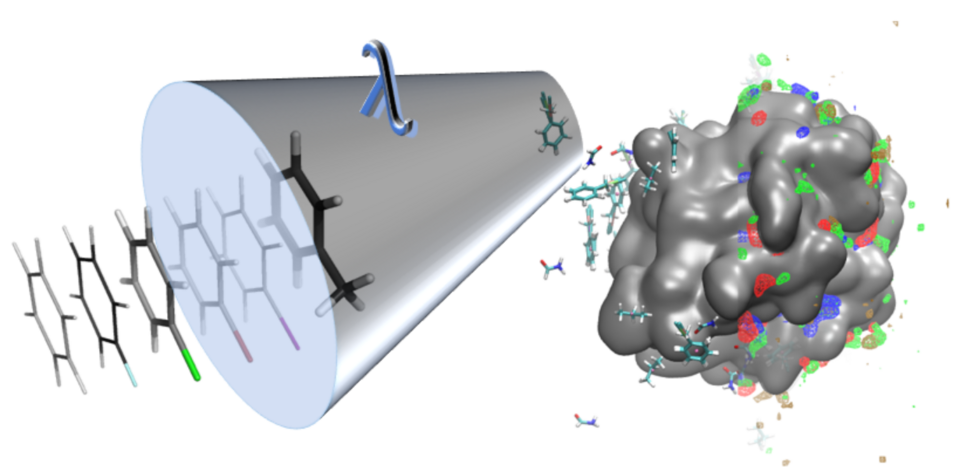
Mahmoud, A. H.; Yang, Y.; Lill, M. A. Improving Atom-Type Diversity and Sampling in Cosolvent Simulations Using λ-Dynamics. J. Chem. Theory Comput. 15, 2019, 3272-3287.
In addition to side-chain and small-backbone flexibility, loop and tail regions of proteins present important flexible moieties critical for ligand or protein binding, and for the function of a protein. Often flexible loops are not isolated but in close proximity to other loop regions. To simultaneously predict the conformations of multiple interacting loop regions critical for ligand binding, we developed a new methodology, titled CorLps. First, an ensemble of individual loop conformations is generated for each loop region. The members of the individual ensembles are combined and are accepted or rejected based on a steric clash filter. After a subsequent side-chain optimization step, the resulting conformations of the interacting loops are ranked by a statistical scoring function. Correlated loops are identified in many important drug targets (e.g. GPCRs and kinases) and off-targets (e.g. cytochrome P450 enzymes). We also studied the influence of ligand binding to the stabilization of alternative loop conformations.
Danielson, M.L.; Lill, M.A. Predicting flexible loop regions that interact with ligands: The challenge of accurate scoring. Proteins, 80, 2012, 246-260.
Danielson, M.L.; Lill, M.A. New computational method for prediction of interacting protein-loop regions. Proteins 78, 2010, 1748-1759.
Improved multidimensional QSAR method
For the accurate quantification of protein-ligand interactions without the knowledge of any protein structure data, we have developed a computational technology, Raptor, which correlates physicochemical properties of the ligands binding to the same protein with their associated binding affinity (quantitative structure-activity relationships, QSAR). The uniqueness of this approach is its simulation of adapting physicochemical properties of the binding site triggered by the ligand binding. It further uses a dual-shell representation of the binding site allowing simulation of various protein substates adopted by different compounds binding to it. The algorithm has been proven to provide realistic 3D binding-site models of the protein and to accurately predict the affinities for sets of structurally similar as well as structurally diverse ligands. By virtue of the accuracy of Raptor in quantifying protein-ligand interactions, we frequently combine docking for generating ligand alignment with subsequent prediction of binding affinities using Raptor. This unique procedure has been successfully applied to nuclear receptors, GPCRs and cytochrome P450 enzymes.
Lill, M.A.; Vedani, A.; Dobler, M. Raptor – combining dual-shell representation, induced-fit simulation and hydrophobicity scoring in receptor modeling: Application towards the simulation of structurally diverse ligand sets. J. Med. Chem. 47, 2004, 6174-6186.